ПЕРСПЕКТИВНЫЕ ИССЛЕДОВАНИЯ
ГЕНЕТИЧЕСКИХ МАКРОМОЛЕКУЛ
Группа научно-образовательного центра «Генетические
макромолекулы: компьютерный анализ и моделирование»,
возглавляемая членом-корреспондентом РАН, д.б.н., профессором
Н. Колчановым, (в составе: к.б.н., В. Иванисенко, к.ф.-м.н.,
И. Титов, к.б.н. Д. Афонников, к.б.н. Ю. Матушкин, Ph.D.
И. Кузнецов, С. Николаев, аспирант С. Пинтус, магистрант
А. Пальянов) выполняет перспективный проект, финансируемый по
грантам международных и российских научных фондов.
Исследования проводятся по следующим направлениям: разработка
методов и алгоритмов предсказания контактов остатков в белках с
использованием информации о паттернах аминокислотных замен;
компьютерный анализ структуры, функции и эволюции активных сайтов
белков; анализ сайтов ДНК-белковых и белок-белковых
взаимодействий; компьютерный дизайн и моделирование искусственных
структур биополимеров.
В рамках выполнения проекта проводятся ежемесячные заседания
семинара группы с обсуждением фундаментальных вопросов
моделирования и анализа РНК и белков, а также с подведением
итогов текущей работы.
Участники проекта занимаются преподавательской деятельностью на
кафедре информационной биологии Факультета естественных наук
Новосибирского государственного университета
(http://www.bionet.nsc.ru/chair/cib/).
Предлагаемый вниманию читателей «НВС» материал подготовлен
участниками проекта и раскрывает основные этапы работ по
выполнению проекта.
Актуальность исследований
Завершение полной расшифровки последовательности генома человека
привело к возникновению новой области науки — системной
биологии. В основе системной биологии лежит моделирование
поведения сложных биологических процессов с целью предсказания их
дальнейшего поведения. Одним из таких фундаментальных процессов
является функционирование живой клетки. Согласно современным
представлениям, наиболее адекватным описанием таких процессов
являются функциональные сети — модели ансамблей
взаимодействующих молекул. Для описания процесса функционирования
различных генов в клетке используют генетические сети. Упрощенно
генную сеть можно представить в виде многочисленных молекул,
которые взаимодействуют друг с другом, обеспечивая, в конечном
счете, контроль активности структурных генов и тем самым
формирование признаков организма. Для описания биохимических
реакций, происходящих в клетке, используют модели метаболических
сетей. Особенностью этих моделей является их сложность, которая
может быть описана в иерархическом виде. На нижних уровнях
иерархии находятся генетические макромолекулы — ДНК, РНК и
белки, а взаимодействия между ними определяют структуру
функциональных сетей. Полное и адекватное исследование функций
генома невозможно без детального исследования всех уровней его
иерархии. Таким образом, в пост-геномную эру на передний план
выдвигаются задачи определения структур генетических
макромолекул, их функций, а также взаимодействий с другими
участниками генных и метаболических сетей.
РНК и белки являются важными генетическими макромолекулами,
которые участвуют во всех процессах жизнедеятельности организмов.
Структура этих молекул имеет иерархическую организацию и включает
первичную структуру (последовательность мономеров), вторичную
структуру (локально упорядоченные участки полимерной цепи) и
третичную структуру (пространственное расположение атомов).
Пространственная структура определяет функциональные особенности
макромолекул и формируется в процессе самосборки. Исследование
закономерностей, определяющих пространственную структуру РНК и
белков, и предсказание их пространственных структур является
одной из важнейших задач биологии на пути к расшифровке механизма
функционирования геномов. Задачей максимум для научного
сообщества в этом направлении является расшифровка структуры всех
белков, последовательности которых секвенированы. С одной
стороны, эта работа будет проводиться экспериментальными методами
(рентгеноструктурный анализ и ЯМР), с другой стороны, часть
результатов может быть получена теоретическими и компьютерными
методами предсказания пространственных структур белков. Развитие
теоретических методов актуально в силу огромных массивов
информации по первичным последовательностям, которые необходимо
обработать для решения этой задачи.
Предсказание структур белков
В ходе выполнения настоящего проекта будут решаться задачи,
связанные с предсказанием таких характеристик структур белков,
как контактные числа аминокислотных остатков и карты контактов
аминокислот в белках. Для этого будут разработаны методы
статистического анализа и предсказания контактов аминокислотных
остатков в белках. Полученные оценки могут быть использованы в
дальнейшем для предсказания пространственных структур белков. На
первом этапе работы был разработан метод предсказания
координационных чисел остатков в белках по аминокислотной
последовательности на основе нейронных сетей. Этот метод
позволяет оценивать конкретное координационное число для позиции
аминокислотной последовательности. В работе также предложено
дополнительно оценивать числа ближних контактов остатка (с
соседними по аминокислотной последовательности остатками).
Показано, что средняя абсолютная ошибка метода составила 4.17 и
1.93 для полного числа контактов и числа ближних контактов,
соответственно. Это, соответственно, на 10% и 15% лучше, чем
предсказание базовым методом, который присваивает каждому типу
аминокислоты значение наиболее часто встречающегося
координационного числа.
Анализ пространственных структур, функции и эволюции
активных
сайтов белков
Известно, что функция белка как объекта генной сети определяется
как его взаимодействиями с другими объектами этой сети (локальная
функция белка), так и его ролью, которую данный белок играет на
уровне всей генной сети (интегральная функция белка). Поскольку
взаимодействие белка с другими компонентами генной сети
обуславливается набором его активных сайтов, то изучение
локальной функции белка невозможно без изучения работы его
активных сайтов. Информация об активных сайтах белков находится в
фокусе разработанной нами базы данных PDBSite. Каждая запись базы
PDBSite соответствует конкретному функциональному сайту
конкретной белковой структуры. Для активных сайтов белков также
рассчитываются их структурные и физико-химические характеристики
(средние значения, сумма и пространственный момент ряда
физико-химических свойств, площадь поверхности, доступной
растворителю, координаты центра масс), показатель разрывности по
первичной структуре, и др. В настоящее время в базе данных
PDBSite аннотировано более 4 тысяч сайтов. PDBSite позволяет
систематизировать информацию об активных сайтах белковых
последовательностей, однако ее анализ требует развития новых
методов. Для этого в ходе выполнения проекта будут разработаны
программы структурного выравнивания сайтов из базы PDBSite с
пространственными структурами белков с учетом особенностей
функциональных сайтов в белковых глобулах. Это позволит получить
закономерности, связывающие структурно-функциональную организацию
сайтов и их пространственное окружение. Будет разработана
структурная классификация функциональных сайтов и новые подходы к
их распознаванию в третичных структурах белков.
 |
 |
| (a) |
(b) |
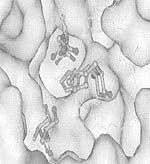
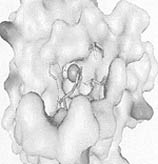
На рисунке показан поиск сайтов с помощью программы PDBSiteScan:
(a) вид выровненных пространственных структур каталитического
центра белка 1ELV и каталитического сайта из базы PDBSite
(идентификатор 1BQYB), демонстрирующий результат работы
программы. Аминокислотные остатки каталитического центра белка
1ELV показаны в виде стержневой модели, аминокислотные остатки
сайта 1BQYB в виде шаростержневой модели;
(b) вид цинк-связывающего сайта ДНК связывающего домена белка p53
человека (идентификатор PDB 1gzh): тонкими линиями показаны
остатки цинк связывающего сайта белка p53; в виде стержневой
модели показаны аминокислотные остатки предположительного участка
связывания цинка, который может формироваться в результате
аминокислотной замены G245аC. Стрелки указывают направление
смещения остатков необходимого для связывания дополнительного
иона цинка.
Особенностью функциональных сетей живых организмов является то,
что они есть продукты эволюции. На уровне генетических
макромолекул эволюция проявляется в стохастическом изменении
последовательности нуклеотидов ДНК. Это приводит к изменению
последовательности и структуры РНК и белков, которые кодируются
геномом. Сравнительный анализ последовательностей и структур
родственных белков позволяет выявлять наборы допустимых замен
аминокислот и оценивать влияние мутаций на структуру и функцию
белка. В ходе настоящего проекта будет проведен анализ
аминокислотных замен в районах активных сайтов белков.
Предварительный анализ показал, что в пространственных структурах
существуют участки, которые имеют расположения атомов, сходные с
расположением атомов в известных сайтах, но отличающиеся
аминокислотным составом на одну аминокислоту. Выдвинута гипотеза,
что такие участки могут формировать новые активные сайты белка в
результате единичной замены аминокислоты. Будет проведен
дальнейший анализ таких замен, произошедших в результате
единичных замен нуклеотидов ДНК (SNP). Будут исследованы
особенности эволюционного процесса и его возможное влияние на
структурные изменения в активных сайтах белков.
Анализ ДНК-белковых и белок-белковых взаимодействий
Функционирование генных и метаболических сетей определяется
взаимодействиями макромолекул. Одними из важнейших являются
ДНК-белковые взаимодействия и белок-белковые взаимодействия.
ДНК-белковые взаимодействия в основном определяют
функционирование генных сетей через механизм регуляции экспрессии
генов. Мы предполагаем провести структурную классификацию
транскрипционных факторов и разработать новые подходы их
распознавания. Наравне с анализом сайтов связывания с ДНК мы
также планируем провести анализ сайтов белок-белковых
взаимодействий. Эта задача актуальна для анализа и предсказания
белковой части генома — протеома. Планируется провести анализ
эволюционных событий в районах сайтов белок-белковых
взаимодействий. Предварительно проведен анализ замен остатков в
альфа-субъединице комплекса протеасомы и проведена классификация
позиций по степени их влияния на эволюционные различия в районах
сайтов белок-белковых взаимодействий.
Компьютерный дизайн и моделирование
искусственных структур
биополимеров
Другой актуальной задачей в пост-геномную эру является дизайн
молекул, обладающих определенными свойствами. Эта задача — одна
из основных в области фармако-геномики. В ходе проекта
предполагается разработка алгоритмов дизайна генетических
макромолекул и контроля за их укладкой в пространственную
структуру на примере решеточных моделей белков. Будет исследовано
влияние внешних воздействий и топологии белков на кинетику
укладки, кинетика заузливания решеточного белка и выявлены
факторы, определяющие скорость и робастность заузливания.
Предварительные расчеты позволили оценить форму поверхности
свободной энергии такого белка и влияние замен аминокислот на
скорость его укладки.
Будут также разработаны подходы к решению обратной задачи для
РНК — предсказанию последовательностей, формирующих заданную
вторичную структуру. Будет разработан подход для восстановления
последовательности РНК по вторичной структуре и рассчитаны РНК,
способные образовывать куб и квадратную решетку.
Анализ эволюции прионовых белков
Одними из интереснейших объектов молекулярной биологии, открытых
совсем недавно, являются прионовые белки. Эти белки, в случае
неправильной сборки, формируют изомер, который нерастворим в
воде, образует нитчатые тяжи и бляшки в пораженных клетках. Такие
изомеры при попадании в нормальную клетку могут вызывать
нарушение ее работы и приводить к развитию нейродегенеративных
заболеваний таких, как скрэпи, болезнь коровьего бешенства и
синдром Крoйцфельда-Якоба.
В 1997 г. Игорь Кузнецов с соавторами провели первый в своем роде
анализ мутационных спектров прионовых белков (Kuznetsov I.B.,
Morozov P. S., Yu.G.Matushkin. Prion proteins: evolution and
preservation of secondary structure. FEBS Letters, 1997, 412,
p.429-432.). Проведенный анализ показал, что, хотя по темпам
эволюции прионовые белки не являются очень консервативными, в
предполагаемых спиральных участках PrP в ходе эволюции
происходили консервативные замены, приводящие к появлению
аминокислот только с очень близкими физико-химическими
параметрами, что свидетельствует о действии отрицательного
отбора, направленного на поддержание вторичной структуры белка.
Применение методов предсказания вторичной структуры белка для
анализа полного спектра одноударных аминокислотных замен в
последовательностях PrP человека показало, что в прионовых
белках, связанных с заболеванием, наблюдается выраженная
тенденция к возникновению замен, нарушающих a-спиральность.
Полученные данные подтверждают предположение о том, что в основе
прионовых заболеваний лежит изменение конформации PrP с
разрушением a-спиралей и образованием b-структур.
И. Кузнецов разработал новый вычислительный алгоритм и с его
помощью идентифицировал в прионовых белках фрагменты, которые
обладают необычными структурными свойствами. Эти фрагменты
являются короткими пептидами с необычайно большой конформационной
гибкостью и обладают так называемыми «свойствами хамелеона». Они
могут принимать либо альфа-спиральную, либо бета-структурную
конформацию в зависимости от белкового окружения. И. Кузнецов
также показал, что остатки, мутации которых вызывают прионовые
заболевания, в структуре прионового белка человека формируют
статистически значимый кластер в районе спиралей B и C [6]. С
помощью анализа профилей последовательностей прионовых белков он
идентифицировал белок UL9 цитомегаловируса шимпанзе, который
может служить структурным шаблоном для моделирования патогенной
конформации прионового белка [7].
Публикации группы за 2003-2004 годы
1. Ivanisenko V. A., Pintus S. S., Grigorovich D. A.,
Ivanisenko L. N., Debelov V. A., Matsokin A. M. PDBSITESCAN: a program
searching for functional sites in protein 3D structures. In:
Bioinformatics of genome regulation and structure. Ed.
By N. Kolchanov and R. Hofestaedt, Kluwer Academic Publishers,
Boston/Dordrecht/London, 2004, pp. 185-192.
2. Afonnikov D. A. Contribution of coordinated substitutions to
the constancy of physicochemical properties of ATP-binding sites
in protein kinases. Bioinformatics of genome regulation and
structure. Ed. By N. Kolchanov and R. Hofestaedt, Kluwer
Academic Publishers, Boston/Dordrecht/London, 2004, pp. 223-230.
3. Н. А. Колчанов, О.А Подколодная, Е. А. Ананько, Д. А. Афонников,
О. В. Вишневский, Д. В. Воробьев, Е. В. Игнатьева, В. Г. Левицкий,
В. А. Лихошвай, Н. А. Омельянчук, Н. Л. Подколодный, А. В. Ратушный
GENEEXPRESS — интегрированная компьютерная система по регуляции
экспрессии генов эукариот, Молек. Биол, 2003, в печати.
4. Афонников Д. А. Предсказание координационных чисел
аминокислотных остатков в белках методом нейронных сетей.
Материалы III конференции молодых ученых, посвященной
М. А. Лаврентьеву, ч. II, Новосибирск, РИЦ «Прайс-Курьер», 2003,
c.43-48.
5. Titov I. I., Palyanov A. Yu. A Genetic Algorithm for The Inverse
Folding of RNA. In: Bioinformatics of genome regulation and
structure. Ed. By N. Kolchanov and R. Hofestaedt, Kluwer Academic
Publishers, Boston/Dordrecht/London, 2004, pp. 193-202.
6. I. Kuznetsov and S. Rackovsky, 2003, Identification of
non-random patterns in structural and mutational data: the case
of prion protein. Proceedings of the IEEE Bioinformatics
Conference. IEEE Computer Society Press. p.604-608.
7. I. Kuznetsov and S. Rackovsky, 2003, Similarity between the
C-terminal domain of the prion protein and chimpanzee
cytomegalovirus glycoprotein UL9. Protein Engineering, 16(12):
861-863.
стр. 7
|
