КАК КОМПЬЮТЕР ПОМОГАЕТ РЕШАТЬ ЗАДАЧИ ПРОТЕОМИКИ
На заседании Президиума СО РАН в начале декабря 2004 г. был
заслушан научный доклад лауреатов Лаврентьевского конкурса
молодых ученых Сибирского отделения — сотрудников Института
цитологии и генетики Д. Афонникова, В. Иванисенко, И. Титова
«Компьютерный анализ и моделирование структуры функций и эволюции
глобулярных белков». Сообщение произвело на собравшихся самое
благоприятное впечатление. Авторы подготовили вариант доклада для
«НВС», который мы и предлагаем читателям.
 |
| Сотрудники научной группы Д. Григорович, А. Пальянов,
С. Пинтус, И. Титов, Д. Афонников, В. Иванисенко. |
Расшифровка последовательностей полных геномов живых организмов
началась в 90-х годах XX века. В 1995 году впервые был
опубликован полный геном свободно живущего организма — бактерии,
вызывающей пневмонию, Haemophilus influenzae. Спустя всего десять
лет стали известны последовательности геномов еще 231 организмов.
Среди них бактерии, грибы, растения, низшие беспозвоночные,
насекомые, рыбы, птицы, млекопитающие и человек. В ближайшие
несколько лет ожидается расшифровка еще почти тысячи полных
геномов.
Эти достижения ставят перед биологией XXI века новые задачи: от
расшифровки нуклеотидных последовательностей геномов необходимо
перейти к определению функции генов и их продуктов — РНК и
белков. Для их решения на стыке биологии, физики, химии,
математики и информатики возникла новая наука — системная
биология. Она изучает закономерности функционирования живых
систем. Практическая цель науки — создание медицины будущего,
которая учитывает индивидуальные генетические особенности каждого
пациента и обладает лекарствами, разработанными на основе
фундаментального знания о взаимодействиях всех объектов живых
систем — ДНК, РНК, белков и органических соединений.
Важная область системной биологии — протеомика — наука о белках
и их роли в живых системах (протеом — набор всех белков,
кодируемых в геноме). Белки способны выполнять специфические
функции благодаря уникальной пространственной структуре, которая
определяется последовательностью аминокислот в полипептидной
цепи. Функции белка обеспечиваются активными сайтами — участками
структуры белка, которые связываются с другими молекулами. Особую
роль в функционировании белка играют взаимодействия с другими
белками, а также с ДНК или РНК. Основные задачи протеомики — определение
состава протеома, пространственной структуры и
функции всех его белков. Решение этих задач позволит
целенаправленно разрабатывать новые лекарственные препараты и
искать для них мишени, создавать белки, обладающие новыми
полезными медицинскими и биотехнологическими свойствами.
В Сибирском отделении РАН исследования в области протеомики
ведутся в ряде научно-исследовательских институтов, а также в
НГУ. Одно из перспективных направлений исследований — компьютерный
анализ и моделирование белков, который проводится в
лаборатории теоретической генетики ИЦиГ, возглавляемой чл.-корр.
РАН Н. Колчановым. Исследования ведутся группой ученых в составе
к.ф.-м.н. И. Титова, к.б.н. Д. Афонникова, к.б.н. В. Иванисенко,
Д. Григоровича, аспирантов С. Пинтуса (НГУ, кафедра
информационной биологии) и А.Пальянова (Институт теплофизики).
За последние три года сотрудниками этой группы при поддержке
молодежных грантов СО РАН и Научно-образовательного центра НГУ
«Молекулярный дизайн и экологически безопасные технологии»,
руководитель которого — акад. В. Болдырев, разработаны
уникальные компьютерные методы и программы. Эти программы
позволяют решать такие актуальные задачи протеомики, как изучение
процесса самосборки белков, предсказание их пространственных
структур, а также анализ и распознавание активных сайтов в
пространственных структурах белков.
Компьютерные модели белковых структур открывают новые возможности
в понимании механизма укладки белка.
Полипептидная цепь принимает уникальную пространственную
структуру в процессе самосборки: белок из развернутой цепи, минуя
множество промежуточных структур, приобретает наиболее стабильную
упаковку, нативную структуру белка. Решение задачи об укладке
полипептидной цепи позволит узнать, как последовательность белка
кодирует его структуру, позволит управлять процессом сборки белка
и синтезировать белки, обладающие заранее заданными свойствами.
Компьютер помогает в решении задачи при использовании численного
моделирования укладки белка. При этом структура белка задается
набором конформационных параметров, а процесс самосборки
представляется как движение точки в многомерном пространстве этих
параметров по поверхности свободной энергии.
Однако описания типичного белка на атомном уровне требует задание
десятков тысяч параметров, и пока не существует компьютеров,
позволяющих решать задачу укладки от начала до конца в атомном
приближении. Ее можно упростить, огрубив геометрию белка или даже
сведя ее к решеточной. В последнем случае аминокислоты
представляются сферами, положения центров которых в пространстве
определяются узлами решетки (обычно кубической); задается энергия
взаимодействия аминокислот, которая зависит от наличия контакта
между ними. На решетке полипептидная цепь может совершать
движения по законам тепловых флуктуаций.
Подобные модели можно исследовать даже на обычном персональном
компьютере, изучая динамику самосборки белка от развернутой цепи
до окончательного формирования нативной структуры. Такие
исследования успешно проводятся в лаборатории старшим научным
сотрудником И. Титовым и аспирантом А. Пальяновым. Ими
исследованы детерминанты укладки решеточного белка, состоящего из
27 звеньев. Оказалось, что наиболее важный фактор быстрой укладки
— компактность структуры и отсутствие стабильных конкурирующих
конформаций. Анализ модели показал, что критическим этапом в
процессе самосборки является формирование структурного ядра
белка, которое ведет к быстрому достижению полипептидной цепью
своего нативного состояния. Было показано, что для ускорения
процесса укладки белка необходимо увеличить скорость формирования
структурного ядра, стабилизируя его при помощи лигандов или
мутациями. Исследование структурных детерминантов укладки белков
и механизмов управления ею получило развитие в следующей работе.
Решение задач укладки белка стало особенно актуально с точки
зрения современной медицины. Совсем недавно был открыт механизм
возникновения ряда загадочных заболеваний, связанных с нарушением
процесса самосборки. Эти нейродегенеративные заболевания, к
которым относятся болезнь коровьего бешенства, болезнь Куру,
поражающая племена каннибалов в Центральной Африке, болезнь
Альцгеймера, даже стали называть общим термином «болезни
неправильной сборки белка». За одно из таких открытий в 1998 году
американцу Стэнли Прусинеру вручили Нобелевскую премию. Прусинер
выяснил, что в нервной ткани человека синтезируется загадочный
белок — прион, функция которого пока остается неизвестной. Он
может принимать пространственные структуры двух типов, одна из
них — глобулярная и водорастворимая, а другая — складчатая и
нерастворимая.
Нерастворимая форма является патогенной: при контакте с
растворимой формой она переводит ее в нерастворимую.
Нерастворимые белки в клетках образуют агрегаты в виде бляшек,
которые приводят к деградации нервной ткани. Формирование
патогенной структуры происходит как в результате внешних
воздействий (изменение кислотности среды), так и в результате
мутаций, однако точные причины остаются до конца неизвестными.
Поэтому возникает актуальная задача исследования конформационных
переходов прионоподобных белков. Это исследование было проведено
И. Титовым и А. Пальяновым совместно с проф. С. Чекмаревым (ИТ) и
коллегами из Института молекулярной биологии (Страсбург) для
решеточной модели 27-мерного белка, имеющего наравне с
низкоэнергетичной нативной структурой альтернативную, энергия
которой лишь немного выше нативной.
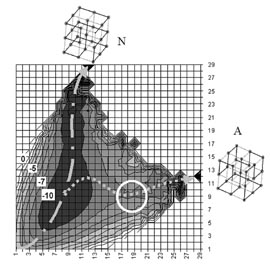 |
|
Рисунок 1.
Поверхность свободной энергии для решеточного белка, который
имеет две низкоэнергетических пространственных структуры,
нативную (N) и альтенативную (A). Пространственные структуры
показаны на рисунке вверху (нативная) и справа (альтернативная).
Координатами на графике являются число контактов, наблюдаемых в
альтернативной структуре (по горизонтали) и в нативной структуре
(по вертикали). Цифрами показаны значения уровней свободной
энергии. Штрих-пунктирной линией показана долина, ведущая к
нативной структуре, пунктирной линией — долина, ведущая к
альтернативной структуре. Энергетический барьер, разделяющий две
долины и препятствующий формированию альтернативной структуры в
обычных условиях, обведен белым кружком.
|
На компьютере была рассчитана поверхность свободной энергии для
такого белка, и оказалось, что она имеет вид двух долин
(рисунок 1). Одна из долин ведет от полностью развернутого состояния к
нативной структуре белка, движение по ней быстро приводит к
нативному состоянию. Другая долина начинается в области
полукомпактных структур и отделена от первой перевалом,
преодолеть который в нормальных условиях белок не в состоянии.
Оказалось, что можно заставить белок перейти в альтернативную
структуру, изменяя высоту перевала при помощи мутаций или
действием лиганда. Таким образом, исследование факторов,
контролирующих укладку белков, позволило (пока еще на
компьютерных моделях) управлять процессом укладки белка.
Компьютерный анализ эволюции позволяет предсказывать структуру
белка.
Пространственные структуры белков в настоящее время известны лишь
для нескольких тысяч белков, в то время как последовательностей
белков известно несколько сотен тысяч. Поэтому компьютерное
предсказание пространственной структуры белка по его
аминокислотной последовательности — одна из важнейших задач
протеомики. Предсказать структуру полипептидной цепи,
оказывается, можно не только с помощью моделирования процесса
укладки, иногда гораздо быстрее и точнее ее можно рассчитать с
помощью информации о последовательностях и структурах родственных
белков с использованием эволюционного анализа. Один из интересных
подходов к решению этой задачи, предсказание пространственной
близости (контактов) аминокислот в структуре белка,
разрабатывается в лаборатории теоретической генетики. Зная наборы
контактов аминокислот, можно довольно точно предсказывать
расположение полипептидной цепи в пространстве. Для предсказания
контактов используется эволюционная информация о координированных
мутациях в белках. Это замены одной или нескольких аминокислот,
которые в ходе эволюции происходят взаимозависимо.
Такие замены обусловлены взаимодействиями аминокислот, и часто
имеют компенсаторную природу, т.е. дестабилизирующий эффект одной
мутации может быть скомпенсирован заменой другой аминокислоты.
Например, для пар заряженных аминокислот, сближенных в
пространстве, чаще будут встречаться аминокислоты, несущие
разноименные заряды (солевой мостик), чем заряды одного знака,
поскольку замены, которые не изменяют суммарный заряд боковых
групп, более предпочтительны.
В последовательностях современных белков такие замены могут
проявляться в виде статистических корреляций величин
физико-химических свойств аминокислотных остатков (например,
объема или заряда). Пример таких координированных замен можно
наблюдать в последовательностях ДНК-связывающих доменов семейства
гомеодомен в позициях белка 19 и 30. Так у плодовой
мушки-дрозофилы в позиции 19 — отрицательно заряженная
аминокислота, в позиции 30 — положительно, у человека наоборот,
и такие пары разноименно заряженных аминокислот в этих позициях
встречаются довольно часто. Оказалось, что боковые группы этих
аминокислот сближены в пространстве и формируют солевой мостик.
Для автоматического поиска координированных замен Д. Афонниковым
разработана специальная компьютерная программа. С ее помощью
проанализированы более трех сотен последовательностей
гомеодоменов, взятых из разных организмов. В результате
оказалось, что кроме обнаруженной ранее пары аминокислот в
позициях 19 и 30 координированные замены происходят в целой
группе соседних позиций, формирующих область контакта двух
структурных доменов белка. С помощью программы были также
проанализированы последовательности других ДНК-связывающих
белков, относящихся к классу «цинковый палец». В результате
анализа на поверхности белка выявлен кластер аминокислот, для
которых характерен режим компенсаторных замен по отношению к
заряду. Интересно, что аминокислоты из этого кластера формируют
протяженную область, участвующую в связывании с ДНК. Таким
образом, полученные данные позволили сделать вывод, что
координированные замены могут быть использованы для предсказания
близости аминокислот в белках и взаимного расположения
структурных доменов. В настоящее время программы и алгоритмы
совершенствуются с целью получения более точных предсказаний.
Компьютерный анализ активных сайтов белков позволяет
предсказывать их функцию.
Еще одна важная задача протеомики — анализ и предсказание
функции белка. Известно, что функция белка определяется его
активными сайтами, поэтому накопление и систематизация информации
об активных сайтах белков чрезвычайно актуальна. В. Иванисенко,
Д. Григоровичем и С. Пинтусом разработана компьютерная база
данных PDBSite, которая содержит информацию о более чем 12
тысячах активных сайтов белков. Источником информации служат
хорошо документированные пространственные структуры белков.
В базе данных описаны сайты связывания малых молекул, ДНК,
белков, фармакологических препаратов, каталитические центры, а
также сайты ковалентной модификации белков. Для каждого сайта
описаны аминокислотный состав, физико-химические свойства и
структурный шаблон, который используется для поиска сайта в
пространственных структурах белков. Для такого поиска разработана
уникальная компьютерная программа PDBSiteScan, которая в течение
нескольких секунд позволяет идентифицировать активные сайты,
выполняющие самые разные функции. База данных и программа поиска
представляют собой интегрированную компьютерную систему, с
помощью которой можно решать широкий класс задач, имеющих
отношение к функции белка.
С использованием этой системы В. Иванисенко и С. Пинтусом
проведено исследование белка p53 человека. Этот белок в клетке
выполняет важную функцию — взаимодействует с ДНК и запускает
механизм самоуничтожения клетки в случае нарушения ее работы.
Неправильная работа белка вызывает неконтролируемое деление
клеток, приводящее к раку. К дисфункции белка p53 приводят
мутации, которых описано около сотни, однако точный механизм
нарушения функции установлен пока не для всех из них. Одна из
таких мутаций — замена глицина в положении 245 белка p53 на
цистеин. Эта мутация вызывает наследственную предрасположенность
к раку (синдром Ли-Фраумени). Глицин в этом положении находится
вблизи участка связывания иона цинка. В нормальном белке
связывание с ионом цинка стабилизирует структуру белка и
обеспечивает его правильное взаимодействие с ДНК. Компьютерные
модели показали, что в результате мутации в непосредственной
близости от участка связывания цинка возможно возникновение
дополнительного сайта, также обладающего способностью связывать
цинк. В итоге ион цинка может сместиться в область нового сайта,
и в структуре белка могут возникнуть искажения, препятствующие
связыванию с ДНК и нарушающие работу p53.
Уникальность разработанной системы заключается еще и в том, что
она предоставляет возможность рассчитывать модели взаимодействия
белков с другими молекулами, используя шаблоны активных сайтов из
базы данных PDBSite. С помощью такой технологии было проведено
исследование белков вируса гепатита С человека. Этот вирус
попадает в организм путем, сходным с вирусом иммунодефицита
человека и поражает печень. Хроническая форма инфекции длится
10-15 лет и вызывает цирроз печени, провоцирует рак, угнетает
иммунную систему организма. В России число инфицированных
оценивается в 2 млн человек. Среди вирусных белков В. Иванисенко
обнаружил белок NS5А, который согласно компьютерной модели может
вступать во взаимодействие с белком NTF2 человека, отвечающего за
транспорт белков в ядро клетки (рисунок 2а). За счет связывания с
транспортером, вирусный белок может попадать в ядро клетки печени
и нарушать ее функцию на самых фундаментальных уровнях
функционирования. С помощью компьютерных расчетов удалось так же
построить модель возможного взаимодействия вирусного белка NS5А и
белка NFAT человека (рисунок 2б), который активен в клеточном
ядре и участвует в регуляции работы иммунной системы. Построенные
компьютерные модели еще ждут своей экспериментальной проверки,
однако, без сомнения, они проливают свет на возможные механизмы
подавления вирусом клеточного иммунитета. Кроме того, выявленные
при помощи компьютерного анализа взаимодействия белков вируса и
человека могут служить мишенью для фармакологических препаратов.
Разработка препаратов, блокирующих такие взаимодействия, позволит
эффективно предотвращать проникновение вирусных белков в ядро
клетки и их вредное воздействие на регуляцию иммунной системы
человека.
| а) | б) |
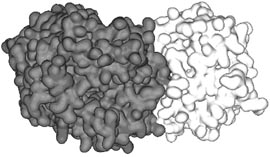 | 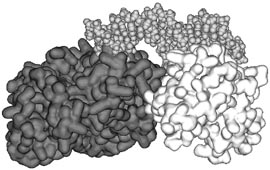 |
Рисунок 2.
Теоретические модели взаимодействия белка NS5A вируса гепатита С
с белками человека, рассчитанные с помощью компьютерной системы
PDBSite/PDBSiteScan.
а) Пространственная модель комплекса вирусного белка, показанного
темно-серым цветом, с белком-транспортером человека NTF2,
показанного светло-серым цветом.
б) Пространственная модель комплекса вирусного белка, показанного
темно-серым цветом, с белком-регулятором иммунной системы
человека NFAT, показанного светло-серым цветом и молекулой ДНК,
показанной серым цветом.
|
Протеомика — новая наука, основные открытия и практические
результаты которой еще впереди. Несомненно, у нее большое
будущее. И оно немыслимо без использования новейших компьютерных
технологий обработки и анализа огромного массива информации,
накопленной в результате экспериментов, численного моделирования
биологических процессов. Важная роль отводится компьютерам и в
развитии новых методов генерации гипотез и их отбора для
дальнейшей экспериментальной проверки. Решение задач протеомики
невозможно без интеграции компьютерных технологий с другими
науками — физикой, математикой, химией. Неслучайно, что работа
группы молодых ученых ведется во взаимодействии с сотрудниками
других институтов СО РАН — проф. С. Чекмаревым (ИТ), проф.
В. Дебеловым (ИВМиМГ СО РАН), группами из Научно образовательного
центра НГУ «Молекулярный дизайн и экологически безопасные
технологии», кафедрой информационной биологии НГУ и зарубежными
коллегами. Это сотрудничество позволит закрепить и развить те
успешные результаты, которые были получены.
стр. 3-4
|
